The UMD-DYSF mutations database
The protein
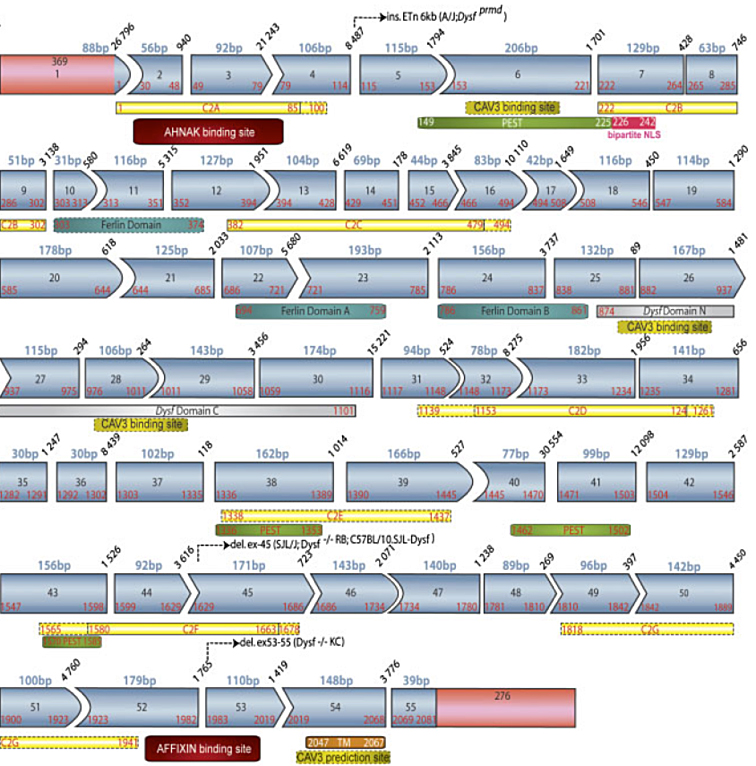
Dysferlin (O75923) is a 237 kDa protein of 2080 amino acids.
Domain organization
Dysferlin is a type II transmembrane protein that belongs to the ferlin family, a protein family composed of dysferlin (FER1L1), otoferlin (FER1L2), myoferlin (FER1L3), FER1L4, FER1L5 and FER1L6.
Structure of the dysferlin sequence consists in seven C2 domains (C2A to C2G, Therrien et al. 2006), a central region of unknown function composed of ferlin family domains and two nested DysF domains, and a carboxy-terminal transmembrane segment that anchors the protein to the membrane.
Coordinates of dysferlin structural domains as annotated in the UMD-DYSF (Show/Hide)
Role and structure of C2 domains (Show/Hide)
Role and structure of DysF domains (Show/Hide)
Tissue distribution and subcellular location
Dysferlin is a ubiquitously expressed protein, with particular high expression in skeletal muscles, cardiomyocytes, placenta and monocytes. It is a major component of the sarcolemma (Anderson et al. 1999), colocalizes with the dihydropyrimidine receptor (DHPR) in T-tubules (Ampong et al. 2005, Klinge et al. 2010) and is also found in cytoplasmic vesicles (Piccolo et al. 2000). It is not an integral component of the dystrophin-glycoprotein complex.
Involvement in disease
The dysferlin gene was first identified as the genetic causes of Miyoshi myopathy and limb girdle muscular dystrophy type 2B (LGMD2B) in 1998 (Bashir et al. 1998, Liu et al. 1998). Since then, other phenotypes caused by mutations in DYSF have been described (see THE CLINICS part). Patients with mutations in the dysferlin gene show severe reduction or complete absence of dysferlin protein in skeletal muscles. In addition, dysferlin-null mice present an accumulation of vesicles at the sarcolemma, near membrane damage sites (Bansal et al. 2003).
Secondary reduction of dysferlin level is also observed in other neuromuscular disorders, including limb girdle muscular dystrophies such as some sarcoglycanopathies (Piccolo et al.
2000), calpainopathies (Chrobakova et
al. 2004) or caveolinopathies (Sugie et al. 2004).
In different forms of muscular dystrophy (dystrophinopathies, sarcoglycanopathies, calpainopathies, fukutin-related protein, and lamin A/C) an altered and predominantly cytoplasmic localization of dysferlin has been described in isolated myofibers (Klinge et al. 2010).
Functional role
Dysferlin deficiency leads to membrane resealing defaults and to severe disruption of the sarcolemma structure (Matsuda et al. 1999). Although its precise biochemical role remains unknown, dysferlin has been shown to play a key role in membrane repair (Bansal et al. 2003) by mediating the fusion of intracellular vesicles to the sarcolemma during two-photons-induced membrane repair in skeletal muscle fibers. Recently, it has been proposed that dysferlin may also be involved in T-tubule formation and maintenance (Klinge et al. 2010).
Mice models for dysferlinopathies
Researchers working on the field of dysferlinopathies have now a plentiful hand of dysferlin deficient models that can be analysed and that can undoubtedly accelerate the pace of dysferlin function studies. Dysferlinopathy profits from two naturally occurring models, the SJL/J and the A/J mice (Bittner et al. 1999, Vafiadaki et al. 2001) and two KO animals (Ho et al. 2004, Bansal et al. 2003).
Detailed information on mice models. (Show/Hide)
Known protein-protein interactions
Dysferlin has been shown to interact with the following proteins:
Alpha-tubulin (TUBA1B, TUBA4A). Polymers of alpha- and beta-tubulins, microtubules are major structural components of eukaryotic cells and are involved in many cellular processes including vesicular transport and protein trafficking to the plasma membrane. Alpha-tubulin interacts with C2A and C2B domains of synaptotagmins I and IX (Honda et al. 2002, Haberman et al. 2003) and it has been suggested that synaptotagmin I interaction with microtubules could retain synaptic vesicles as a readily releasable pool, close to the exocytosis active zone.
Alpha-tubulin also interacts with C2A and C2B domains of dysferlin, in a Ca2+-independent manner (other dysferlin C2 domains were not found to bind alpha-tubulin) (Azakir et al. 2010). In myoblasts, microtubules nucleate at the centrosome and project towards the plasma membrane and co-localization of dysferlin and alpha-tubulin was found in the perinuclear region, as well as in vesicular structures (Azakir et al. 2010). In myotubes, co-localization of dysferlin with alpha-tubulin was found in thin longitudinal structures indicative of microtubules (Azakir et al. 2010). It is thus hypothesized that interaction of dysferlin with microtubules could provide a ready pool of dysferlin for membrane resealing.
Caveolin-3 (CAV3/LGMD1C) is a scaffolding protein within caveolar membranes and T-tubules that belongs to the dystrophin-glycoprotein complex. It regulates the sarcolemmal membrane stability and is involved in vesicular trafficking and signal transduction. Defects in caveolin-3 can be at the origin of several skeletal muscle disease phenotypes: limb girdle muscular dystrophy 1C, rippling muscle disease, distal myopathy and isolated hyperCKaemia (Gazzerro et al. 2010). Caveolin-3 inhibits endocytocis of sarcolemmal dysferlin via a clathrin-independent pathway (Hernandez et al. 2008) and mutations in CAV3 are associated with a mislocalization of dysferlin in muscle cells (Matsuda et al. 2001) with accumulation in the cytoplasm or irregular membrane distribution of dysferlin.
MG53 (TRIM72): MG53 is specifically expressed in striated muscles where it interacts with caveolin-3 to regulate membrane budding and exocytosis by nucleating recruitment of intracellular vesicles to injury sites for membrane patch formation (Cai et al. 2009a, Cai et al. 2009b). MG53 is required for the transport of dysferlin to sites of cell injury during repair patch formation (Cai et al. 2009c).
Annexins A1 and A2 (ANXA1/2) are involved in membrane trafficking and exocytosis, transmembrane channel activity, phospholipase A2 inhibition and cell-matrix interactions (Raynal et al. 1994). Annexins are Ca(2+)-dependent phospholipid binding proteins which promote membrane fusion by aggregating intracellular vesicles and lipid rafts to the cytosolic surface of the plasma membrane. Following membrane injury, the disruption of annexin-dysferlin interaction mediates sarcolemma repair via a Ca(2+)-dependent aggregation and fusion of vesicles to the membrane (Lennon et al. 2003).
Beta-parvin/affixin (PARVB) is an actin and integrin-linked kinase–binding protein that plays a role in the regulation of cell adhesion and cytoskeleton organization. Dysferlin was shown to interact with beta-parvin at the sarcolemma (Matsuda et al. 2005).
AHNAK and AHNAK2 proteins are both localized
in Z-band regions of cardiomyocytes and bind to sarcolemma and T-tubule
membranes. AHNAK is thought to be involved in calcium homeostasis and in signal
transduction at the cell membrane: in
vitro, the AHNAK protein can produce diacylglycerol and inositol
triphosphate by activating phospholipase C (Sekiya et al. 1999).
In addition, predicted 2D-structure of AHNAK2 suggests a 3D-spatial
organization similar to beta-propeller proteins such as RCC1, clathrin and G
beta-subunit and indicates a role of AHNAK proteins as scaffolding proteins (Rosa and
Barbacid 1997, Komuro et al. 2004).
AHNAK interacts with dysferlin C2A domain in a Ca(2+)-independent
manner (Huang
et al. 2007) and this interaction is regulated by the calpain-3 protease
activity (Huang
et al. 2008). Furthermore, in dysferlinopathies, dysferlin deficiency in muscles
correlates with a secondary loss of AHNAK and both proteins show an increased
localization in cytoplasm in rat regenerating muscles (Huang et al. 2007).
Thus it has been hypothesized that dysferlin may recruit AHNAK/AHNAK2 to the
plasma membrane.
Calpain-3 (CAPN3/LGMD2A). One of the most common LGMD2 forms is caused by loss of function mutations in the LGMD2A gene that affect its product calpain-3, a protein that belongs to the calcium-dependant cysteine protease family and that seems to play a central role for remodelling the sarcomeres (Duguez et al. 2006). Calpain-3 interacts with dysferlin (Huang et al. 2005) and its expression is reduced in dysferlin deficient muscle cells and vice versa. Moreover, AHNAK, another dysferlin-interacting protein is a substrate of calpain-3 and accumulates at the sarcolemma in CAPN3 deficient patients (Huang et al. 2008). It is thus proposed that calpain-3 may play an important role in cytoskeleton-membrane interactions. However, calpain-3 have not yet been proved to play a role in the early stage of membrane repair (Mellgren et al. 2009).
Schematic representation of structural and protein-protein interacting domains alongthe protein sequence (Show/Hide)