The DMD mutations database
The dystrophin protein
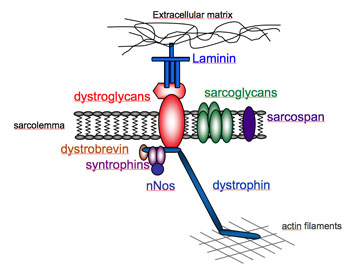
The dystrophin-associated complex
Full-length dystrophin is a large, rod-shaped protein of 427 kDa composed of 3685 amino acid residues. In skeletal muscle fibers, the protein localizes at the cytoplasmic face of the sarcolemma (Watkins et al.), where it constitutes 5% of sarcolemmal protein and 0.002% of total striated muscle protein (Hoffman et al.). Dystrophin is present in normal individuals from fetal life onwards in all skeletal, heart and smooth muscle, and in some neuronal cell types.
At the sarcolemma, dystrophin is part of a macromolecular group of proteins collectively referred to as the dystrophin-associated protein complex (DAP) and include sarcoglycans (α, β, δ, γ, ε), dystroglycan (α, β), dystrobrevins, syntrophins (α, β), sarcospan, caveolin-3, and NO synthase proteins (Lapidos, K.A. et al.). Dystrophin binds to the intracellular cytoskeleton by associating with actin filaments at its N-terminus, whereas at its C-terminus dystrophin interacts with members of the DAP, including β-dystroglycan. On the extracellular side of the membrane, α-dystroglycan, binds to β-dystroglycan and also acts as a receptor for the extracellular-matrix.
Structure of the dystrophin
Based on its primary structure, the dystrophin can be divided into four distinct structural domains: (1) the N-terminal actin-binding domain (aa 14-240), (2) the large triple helical spectrin-like domain (aa 253-3040) composed of 24 repeating units similar to those in β-specrin, which are predicted to form triple-helical coiled-coils, (3) the cysteine-rich domain (aa 3080- 3360) and the C-terminal domain (aa 3361- 3685) (Koenig, M., Monaco, A. P., and Kunkel, L. M.). A second actin binding domain has been identified in the rod region in spectrin repeats 11 through 17. The flexible hinge regions (H1-H4) are proline-rich sequences that interrupt the rod domain. The C-terminal region contains a number of protein domains that form the binding sites for some of the components of the DAP. These are the WW domain (WW), the ZZ domain (ZZ) that is located in the cysteine-rich region (CYS), and the coiled-coil domain (CC) in the C terminus (CT). The 15 C-terminal amino acids of -dystroglycan constituted a unique binding site for the second half of the hinge 4 and the cysteine-rich domain of dystrophin (amino acids 3054-3271) (Jung et al.). The extreme carboxyl-terminal region is α-helical in nature and mediates its interaction with the syntrophins (Suzuki et al.)
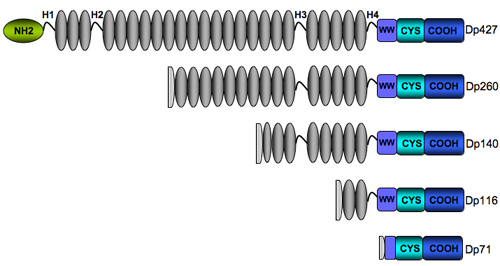
The four shorter dystrophin isoforms retain the cysteine-rich and C-terminal domains of dystrophin, preserving the binding sites for dystroglycan, dystrobrevin, and syntrophin, with various extensions into the spectrin-like repeats domain. They all lack the actin-binding terminus suggesting they may have other functions different from those ascribed to full-length dystrophin. Of the different isoforms, Dp140 and Dp71 have both been associated with an increased risk of mental retardation which suggests a strong presence of these isoforms in the central nervous system (Bardoni et al., Moizard et al., Mehler).
Roles of the dystrophin
The precise molecular roles of dystrophin are still to be elucidated, but primarily it appears to stabilize the sarcolemma and to protect muscle fibres from long-term contraction-induced damage and necrosis (Davies KE, Nowak KJ). Dystrophin serves to link the intracellular microfilament network of actin to a complex series of linking proteins in the cell membrane, and hence to the extracellular matrix (Ervasti and Campbell). It was found that the C-terminus of dystrophin, which binds it to the sarcolemma, is crucial, as is the acting-binding zone, but the helical zone can be shortened with some preservation of function, explaining the differences between DMD and BMD. Nonstructural roles have also been described for dystrophin, making it a multifunctional protein. Dystrophin would act as a scaffold for several signaling molecules, but the roles of dystrophin-mediated signaling pathways remain unknown. (Rando). However, the mounting evidence pointing to a role for the dystrophin complex in localizing signaling mediators argues in favor of the notion this function may be important in stimulating appropriate downstream cascades in response to various extracellular stimuli. Finally, dystrophin has been proposed to play a role in calcium homeostasis (Chakkalakal et al.).