The CFTR mutations database
The CFTR gene
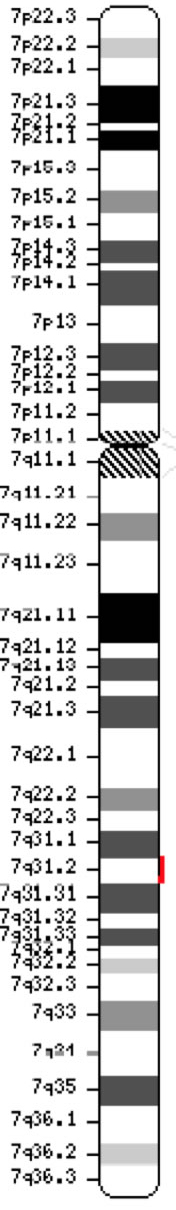 |
The cystic fibrosis transmembrane conductance regulator gene (CFTR, OMIM #602421) is a member of the ATP-binding cassette sub-family C (ABCC7). The gene, discovered in 1989 (Kerem et al, 1989; Riordan et al, 1989; Rommens et al, 1989), spans 188,702 bp on the long arm of chromosome 7 (band 7q31-q32) (position 117,119,358-117,308,719) and is composed of 27 exons initially numbered 1-24 with subdivisions a and b for exons 6, 14 and 17 (Zielenski et al., 1991). The mature transcript is 6,129 nucleotides long including an open reading frame of 4,440 coding bases (Reference sequence: NM_000492). Ensembl Gene ID: ENSG00000001626 |
Nomenclature
The UMD-CFTR uses the recommended nomenclature guidelines proposed by the Human Genome Variation Society (www.hgvs.org/mutnomen/) e.g., cDNA-based numbering with the A of ATG translational initiation codon at +1 and exons numbered 1 to 27.
In the UMD-CFTR the coding reference sequence is NM_000492 with the exception of c.1408A instead of G, and the reference protein sequence is NP_000483 with the exception of p.Met470 instead of p.Val470.
For convenience to the users who are accustomed to the cystic fibrosis mutations database consortium numbering (www.genet.sickkids.on.ca/cftr/) two downloadable files are provided:
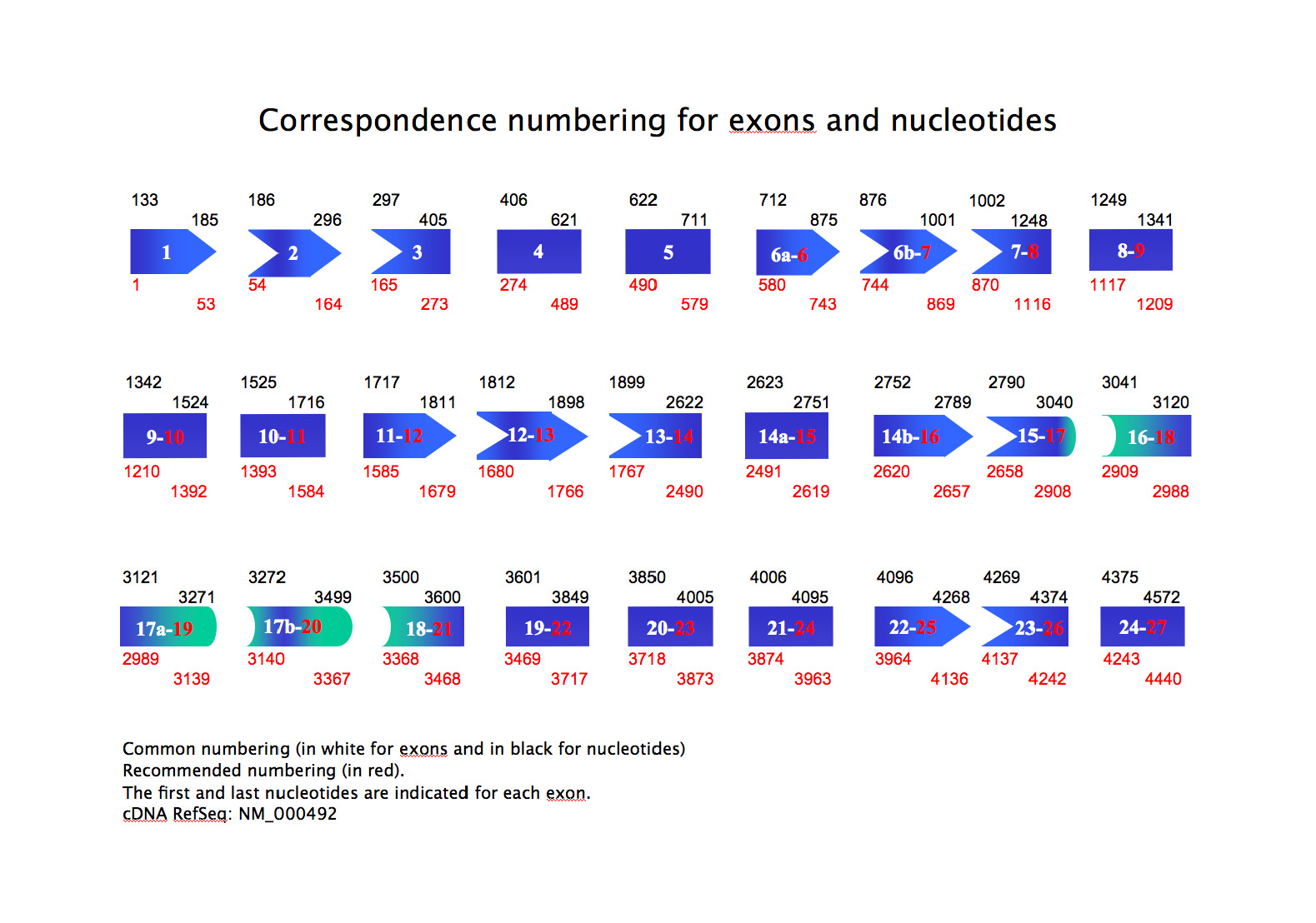
- a correspondence numbering for exons and nucleotides: common numbering (in white for exons and in black for nucleotides) and recommended numbering (in red). The first and last nucleotides are indicated for each exon. (download) (Show/Hide)
- a correspondence table for variation names. (download)
Coding sequence (Show/Hide)

The CFTR mutations
Almost 1,700 CFTR sequence mutations and variations have been described so far (www.genet.sickkids.on.ca/cftr). The most common mutation worldwide responsible for cystic fibrosis is a deletion of three bases encoding a phenylalanine residue at position 508 named p.Phe508del, which impairs the ability of the CFTR protein to fold in the endoplasmic reticulum (ER), thereby enhancing the rapid degradation of the protein during ER processing. It accounts for about 70% of CF alleles.
Heterologous expression of full-length p.Phe508del-CFTR-cDNA has demonstrated that this mutant protein acquires only core glycosylation and not complex oligosaccharide chains and, as such, fails to be transported to the cell surface, in accordance with the observation of absence of mutant protein in fresh epithelia from patient tissues. Among other detrimental effects, p.Phe508del impairs the correct folding of CFTR and also conformationally masks the diacidic exit code of three residues in the NBD1 that seems necessary for ER export.
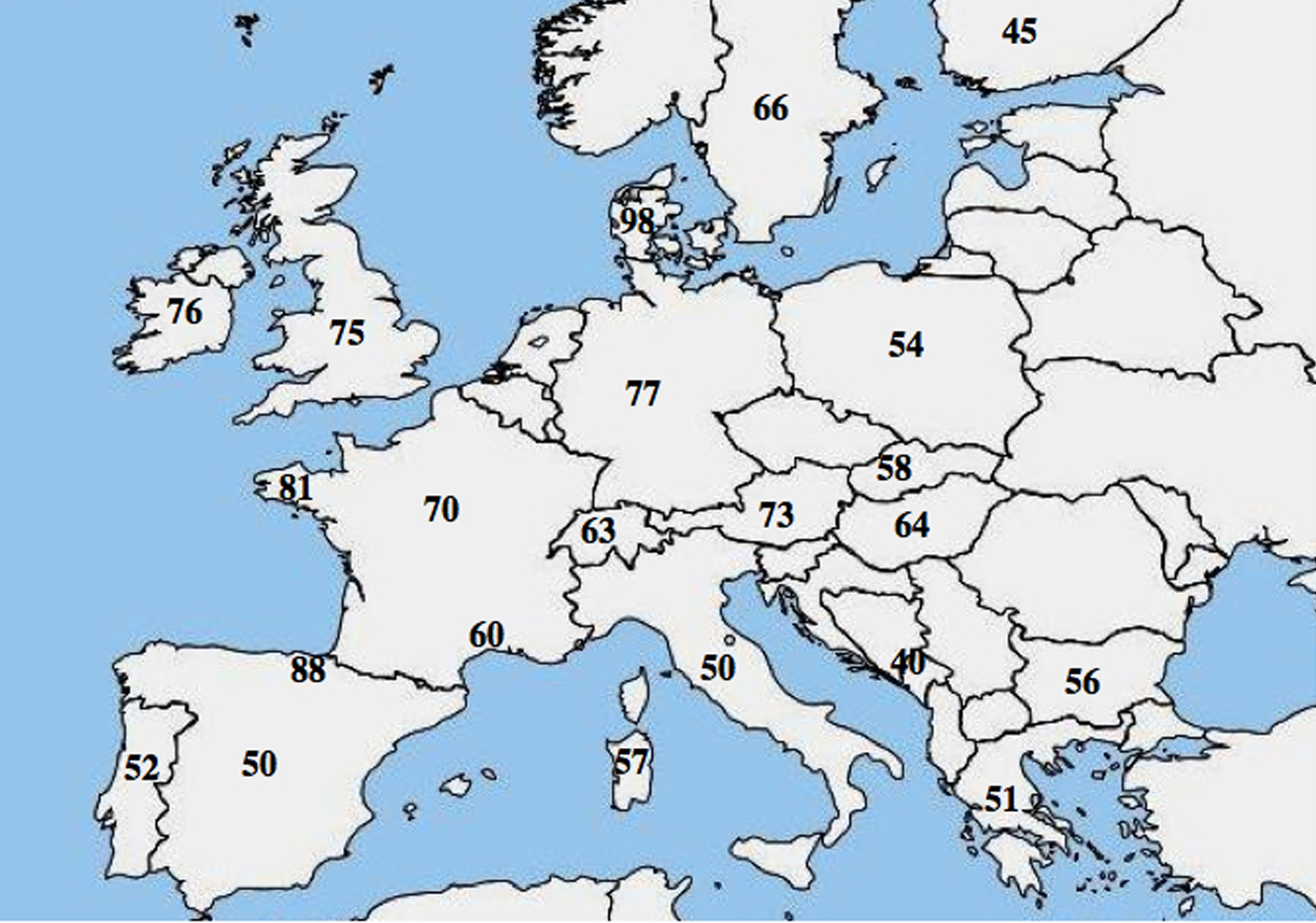
A molecular allelic heterogeneity in CF has been reported; numerous studies highlighted a marked difference in the frequency of p.Phe508del among populations and showed a decreasing northwest to southeast gradient in Europe.
Classes of CFTR mutations
Mutations in the CFTR gene can be classified in six classes according to their molecular mechanisms and consequences for different aspects of CFTR biogenesis, metabolism and function.
Class I: mutations interfering with protein synthesis. They result in the introduction of a premature signal of termination of translation (stop codon) in the mRNA: nonsense, frameshift or severe splicing mutations. The truncated proteins are unstable, rapidly degraded, so, the net effect is no protein at the apical membrane (p.Gly542X, c.489+1G>T (621+1G>T), c.579+1G>T (711+1G>T)). |
Class II: mutations affecting the protein maturation. These lead to the production of a protein that cannot be correctly folded and trafficked to its site of function on the apical membrane (p.Phe508del, p.Ile507del, p.Asn1303Lys). |
Class III: mutations altering the channel regulation. The mutated protein is properly trafficked and localized to the plasma membrane but cannot be activated or function as a chloride channel (missenses located within the NBD), (p.Gly551Asp). |
Class IV: mutations affecting chloride conductance. The CFTR protein is correctly trafficked to the cell membrane but generates reduced Cl- flow (most are missenses located within the membrane-spanning domain), (p.Arg117His, p.Arg334Trp). |
Class V: mutations reducing the level of normally functioning CFTR at the apical membrane (partially aberrant splicing mutation or inefficient trafficking missenses) (c.1210-12T[5] (5T allele), c.3140-26A>G (3272-26A>G), c.3850-2477C>T (3849+10kbC>T)). |
Class VI: mutations decreasing stability of CFTR present or affecting the regulation of other channels. Although they have been investigated in a few patients, most class VI mutants should be considered as severe. |
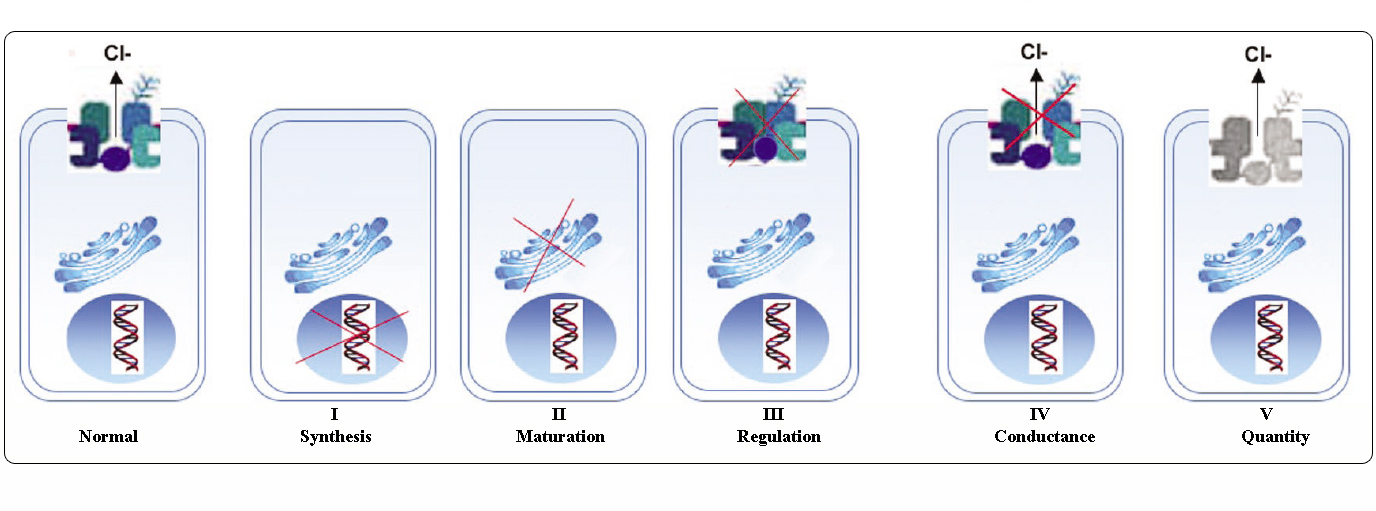
Adapted from Welsh and Smith (1993) and modified from Claustres (RBM online, 2005)
Mutations belonging to classes I-III and VI confer little or no functional CFTR at the apical membrane and are considered as "severe" and usually lead to a classic CF phenotype with pancreatic insufficiency (CF-PI) although the severity of lung disease may be variable.
Mutations belonging to classes IV and V retain some residual CFTR activity and confer a milder phenotype. In patients with at least one "mild" CF allele, CFTR function is usually sufficient for digestion, and the lung disease is less severe. Individuals with CFTR mutants that give rise to about 10% of the normal level of CFTR mRNA may present with only one symptom, such as CBAVD (congenital bilateral absence of vas deferens) in males.
Some individuals heterozygous for a CFTR mutation might be at increased risk for "CFTR-related disorders" (CFTR-RD) such as pancreatitis, sinusitis, or allergic bronchopulmonary aspergillosis. In these cases, CFTR mutations could be a factor of genetic predisposition and genetic or environmental modifiers could play additive effects.
Grouping mutations into different classes is useful for understanding the mechanism of dysfunction, however many sequence variations are rare/unique and available data do not allow to assess their pathogenicity. Moreover, a single mutation can cause more than one type of abnormality or may confer variable phenotype.
References
* Claustres M. 2005. Molecular pathology of the CFTR locus in male infertility. Reprod Biomed Online 10(1):14-41. PMID: 15705292.
* Estivill X. 1996. Complexity in a monogenic disease. Nat Genet 12(4):348-50. PMID: 8630481.
* Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identification of the cystic fibrosis gene: genetic analysis. Science 245(4922):1073-80. PMID: 2570460.
* Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL and others. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245(4922):1066-73. PMID: 2475911.
* Rommens JM, Zengerling-Lentes S, Kerem B, Melmer G, Buchwald M, Tsui LC. 1989. Physical localization of two DNA markers closely linked to the cystic fibrosis locus by pulsed-field gel electrophoresis. Am J Hum Genet 45(6):932-41. PMID: 2589321.
* Welsh MJ, Smith AE. 1993. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73(7):1251-4. PMID: 7686820.
* Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, Rommens J, Tsui LC. 1991. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 10(1):214-28. PMID: 1710598.
* Zielenski J, Patrizio P, Corey M, Handelin B, Markiewicz D, Asch R, Tsui LC. 1995. CFTR gene variant for patients with congenital absence of vas deferens. Am J Hum Genet 57(4):958-60. PMID: 7573058.