The CFTR mutations database
The clinic
Cystic Fibrosis
Cystic Fibrosis (CF) (OMIM #219700), inherited as an autosomal Mendelian recessive trait affects individuals from European descent at a frequency of about 1 in 3,500 live births. It is a life-shortening disorder characterized by the production of abnormally thick and sticky mucus obstructing several organs, leading to progressive chronic and life-threatening lung infections and pancreatic insufficiency due to obstructed ducts and autolysis in about 85% of CF patients. The earliest clinical postnatal manifestation of CF is meconium ileus, which affects about 10-15% of CF babies.
The first clear description of the phenotype goes back to 1938. The most common symptoms of CF include very salty-tasting skin, persistent coughing, wheezing or shortness of breathing, and an excessive appetite but poor weight gain as well as greasy, bulky stools due to malabsorption of fat and protein. Respiratory infections increase with age. The small airways are the primary site of lung disease, with intense inflammation. Decreased secretion of pancreatic enzymes is the main cause of poor growth, fatty diarrhea and deficiency in fat-soluble vitamins. Males are usually azoospermic due to CBAVD.
Discovery of the sweat electrolyte defect in 1948 (patients with CF have a fivefold excess of sodium and chloride excreted in the sweat) and its standardization in 1959 offered a diagnostic test (sweat test), and allowed identification of milder cases. In 1980, new insight on inflammation led to the concept that electrolyte defect was able to contribute per se (independently to infection) to the lung disease. In 1983, chloride transport was identified as the basic physiologic defect in CF (impermeability to Cl- is the hallmark of CF cell), associated with an increased sodium reabsorption. In 1989, the discovery of the CFTR gene demonstrated the basic defect to be in a cAMP-regulated chloride channel and afforded new mutation-based diagnostic tests as well as new therapeutic avenues.
Although severity, age at onset and clinical manifestations may be dependent on other genetic and environmental influences, mutations in the CFTR gene are the primary cause of the disease and virtually all individuals diagnosed clinically with CF have mutations in both CFTR gene alleles. By combining the study of the segregation of mutations and associated haplotypes in a family with a CF case, it is possible to determine the status of a fetus (prenatal diagnosis) or an embryo (preimplantation genetic diagnosis).
Changes in CF care since the early 1980's have contributed to substantial improvements in malnutrition, lung function, and mortality among children and adolescents with CF, so that the median survival age changed from 6 months in 1938 to over 37 years in 2009. The demographics of CF is changing, with almost 50% of patients now older than 18 years. Cystic Fibrosis can now be identified after birth through newborn screening on a blood spot combining quantification of immunoreactive trypsin (elevated in CF) followed by screening for the most common CF mutations. Although this early detection may be controversial because it over-detects CF, it is more and more developed because it should improve the life expectancy and the life quality of patients affected with CF before chronic lung infection is established and, perhaps, avoid fatal issue.
The clinical heterogeneity of CF is mostly explained by the distinct molecular consequences of the mutations that affect the CFTR gene; however other genes as well as environmental exposure appear to modulate the phenotype.
Links
Adult Cystic Fibrosis Committee of Québec (www.acfcq.qc.ca/e/information/information.html)
The corporation’s objectives are as follows: promote, defend and protect the rights and interests of adults with CF in Quebec; promote their quality of life; provide information, represent Quebec’s adult CF population in government organizations and other groups; bring adults with CF together around joint projects geared to the greater welfare of those suffering from the disease or in difficulty.
Asociación Mexicana de Fibrosis Quística (www.fibrosisquistica.org.mx/home/index.php?id=1)
Canadian Cystic Fibrosis Foundation (www.cysticfibrosis.ca/)
The Foundation’s primary objective is to fund cystic fibrosis research and care. The Canadian Cystic Fibrosis Foundation is one of the world’s largest non-governmental granting agencies in the field of cystic fibrosis research.<br>
CF-network (cf.eqascheme.org/)
The European Cystic Fibrosis Thematic Network brought together all parties involved in the same quest "the fight against CF", with the aim of making information more easily available to everybody.
Cystic Fibrosis Foundation (www.cff.org/)
The mission of the Cystic Fibrosis Foundation, a nonprofit donor-supported organization, is to assure the development of the means to cure and control cystic fibrosis and to improve the quality of life for those with the disease.
Cystic Fibrosis Trust (www.cftrust.org.uk/)
Since the founding of the CF Trust in 1964, we have been working to improve the lives of people with CF, raise the profile of CF and fund research into a cure. Our objectives are to: fund medical and scientific research to develop a cure and provide effective treatments for Cystic Fibrosis, ensure appropriate clinical care for those with Cystic Fibrosis, provide information, advice, support and, where appropriate, financial assistance to anyone affected by Cystic Fibrosis.
Cystic Fibrosis Worldwide (www.cfww.org/)
Cystic Fibrosis Worldwide promotes access to knowledge and appropriate care to those people living with the cystic fibrosis and among medical, health professionals and governments worldwide.
European Cystic Fibrosis Society (www.ecfs.eu/)
The European Cystic Fibrosis Society (ECFS) aims to achieve the best possible treatment and the highest quality of life for the patient with cystic fibrosis by the development and distribution of knowledge in the field of cystic fibrosis. The ECFS provides an international forum on all aspects of CF with membership of the society open to all actively engaged in CF research or clinical care.
Vaincre La Mucoviscidose (www.vaincrelamuco.org/ewb_pages/e/english-version.php?lang=en)
“Vaincre la Mucoviscidose” is a non-profit organization. The association is organized around 4 missions: cure CF by helping and financing research in France and all around Europe, treat right now by improving health care, improve quality of life in order to make a life with CF more acceptable and bearable, alert on the gravity of CF by communicating toward the public in general and towards parents and family affected with CF.
References
* Davis PB. 2006. Cystic fibrosis since 1938. Am J Respir Crit Care Med 173(5):475-82. PMID: 16126935. * Kerem E, Conway S, Elborn S, Heijerman H. 2005. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros 4(1):7-26. PMID: 15752677. * Quinton PM. 1983. Chloride impermeability in cystic fibrosis. Nature 301(5899):421-2. PMID: 6823316. * Stern RC. 1997. The diagnosis of cystic fibrosis. N Engl J Med 336(7):487-91. PMID: 9017943. * Correlation between genotype and phenotype in patients with cystic fibrosis. The Cystic Fibrosis Genotype-Phenotype Consortium 1993. N Engl J Med 329(18):1308-13. PMID: 8166795. |
Congenital Bilateral Absence of Vas Deferens (CBAVD)
Congenital Bilateral Absence of the Vas Deferens (OMIM #277180) accounts for about 2% of cases of male infertility and is present in 25% of patients with excretory azoospermia. Diagnosis of isolated CBAVD is based on impalpable vas deferens on scrotal examination associated with total absence of spermatozoa and an acidic ejaculate composed only of prostatic secretions with low pH (<6.8), low volume (<1.5 ml) and low concentrations of fructose (<2 µmol/ejaculate) (Von Eckardstein et al., 2000). CBAVD is regularly accompanied by aplasia of the epididymal tail and body and various anatomical or functional abnormalities of the seminal vesicles.
Because of the striking similarity between the anatomical characteristics and semen parameters for CF and CBAVD, these two disorders were postulated to have a common genetic origin as early as 1968 (Kaplan et al. 1968), but the definite link between CBAVD and CF was proven after the identification of the gene responsible for CF when it was found that a significant proportion of men with isolated CBAVD were heterozygous for the p.Phe508del mutation (Dumur et al., 1990) and for a quantitative CFTR variant in a non-coding intronic DNA sequence (c.1210-12T[5] or 5T allele) (Chillon et al. 1995).
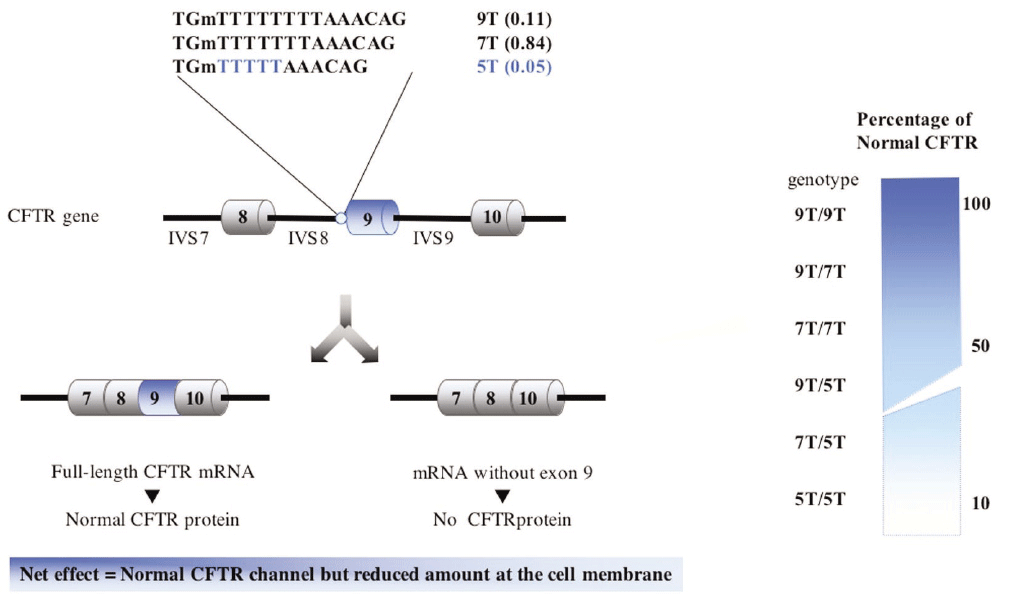
Claustres (RBM online, 2005)
The CFTR exon 10 (usual exon 9) is skipped in human transcripts (Chu et al., 1991) and the degree of exon skipping is inversely correlated with the length of a polymorphic polythymidine tract (Tn) upstream of the exon. In the presence of a T[5] allele, the polypyrimidine tract is too short and under-utilized as a splice acceptor site, resulting in reduced efficiency of exon 10 (usual exon 9) splicing. The CFTR mRNA deleted for exon 10 (usual exon 9) maintains an open reading frame and would encode a CFTR protein that is missing 60 amino acids (position 404 through 464) of the first NBD and, as such, is not properly processed and unable to reach the membrane Delaney et al. 1993).
Extensive analysis of the CFTR gene has demonstrated a distinct spectrum of mutations in CF and in CBAVD (for example, the frequency of p.Phe508del is 21% in CBAVD versus 76% in CF).
Routine testing for the most prevalent mutations in the CF Caucasian populations will miss most CFTR gene alterations in CBAVD, which can be detected only through exhaustive scanning or sequencing of CFTR sequences.
References
* Chillon M, Casals T, Mercier B, Bassas L, Lissens W, Silber S, Romey MC, Ruiz-Romero J, Verlingue C, Claustres M and others. 1995. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med 332(22):1475-80. PMID: 7739684. * Claustres M. 2005. Molecular pathology of the CFTR locus in male infertility. Reprod Biomed Online 10(1):14-41. PMID: 15705292. * Chu CS, Trapnell BC, Murtagh JJ, Jr., Moss J, Dalemans W, Jallat S, Mercenier A, Pavirani A, Lecocq JP, Cutting GR and others. 1991. Variable deletion of exon 9 coding sequences in cystic fibrosis transmembrane conductance regulator gene mRNA transcripts in normal bronchial epithelium. Embo J 10(6):1355-63. PMID: 1709095. * Delaney SJ, Rich DP, Thomson SA, Hargrave MR, Lovelock PK, Welsh MJ, Wainwright BJ. 1993. Cystic fibrosis transmembrane conductance regulator splice variants are not conserved and fail to produce chloride channels. Nat Genet 4(4):426-31. PMID: 7691356. * Dumur V, Gervais R, Rigot JM, Lafitte JJ, Manouvrier S, Biserte J, Mazeman E, Roussel P. 1990. Abnormal distribution of CF delta F508 allele in azoospermic men with congenital aplasia of epididymis and vas deferens. Lancet 336(8713):512. PMID: 1975022. * Kaplan E, Shwachman H, Perlmutter AD, Rule A, Khaw KT, Holsclaw DS. 1968. Reproductive failure in males with cystic fibrosis. N Engl J Med 279(2):65-9. PMID: 5657013. * von Eckardstein S, Cooper TG, Rutscha K, Meschede D, Horst J, Nieschlag E. 2000. Seminal plasma characteristics as indicators of cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in men with obstructive azoospermia. Fertil Steril 73(6):1226-31. PMID: 10856487. |